
Does Radiation Cause Cancer?
The author discusses the multistage aspects of cancer formation and how acute irradiation may affect these processes.
by James E Trosko, RERF Chief of Research
This article was originally published in RERF Update 4(1):3-5, 1992.

Trosko
What an outrageous question! With an enormous amount of experimental animal and human epidemiological data showing that a wide variety of cancers appear after exposure to various kinds of radiation, one could hardly question the conclusion that radiation does “cause” cancer. But wait! Our understanding of carcinogenesis today leads us to believe that it is a multistage, multimechanistic process, involving the interaction of many external and endogenous factors. Consequently, it is misleading to assume that any single factor or “carcinogen”–chemical or physical–“causes” cancer. The key word here is cause.
Carcinogenesis involves many steps and mechanisms, with the interaction of external determinants such as chemical and physical pollutants, medication/drugs, mutagenic and epigenetic agents–as these may occur in the diet or as workplace and environmental pollutants–and endogenous factors related to genetic background, sex, developmental stage, number of stem or progenitor cells that give rise to cancer, DNA repair systems, hormones, growth factors, oncogenes, tumor suppressor genes, and antimetastasis genes.
Carcinogenesis as a multistep, multimechanistic process
Currently, the multistage model of carcinogenesis, derived from whole animal experimental studies, seems to be a plausible model for human carcinogenesis. This model indicates that the first step in carcinogenesis–the initiation stage–is irreversible. The observation that mutagens appear to be effective initiators implicates mutagenesis as the mechanism underlying the initiation stage. The fact that the initiation process appears to be irreversible is also consistent with the hypothesis that mutagenesis is at least one mechanism of initiation. Stable epigenetic repression or activation of genes may be another.
Most cancer studies have been consistent with the clonal theory of cancer, ie, the assumption that cancer arises from changes initiated in one cell (Figure 1). Therefore, the second step in carcinogenesis–the promotion stage–appears to involve the clonal expansion of an initiated stem cell, which, because it is unable to terminally differentiate, accumulates as a focus of nonterminally differentiated cells. Examples of such foci might be papillomas of the skin, enzyme-altered foci of the liver, polyps of the colon, and nodules of the breast. Obviously, this process must require stimulation of cell division (ie, it must be mitogenic), at least with respect to the initiated cell. As demonstrated in experimental animals, this stage is potentially interruptable and reversible.
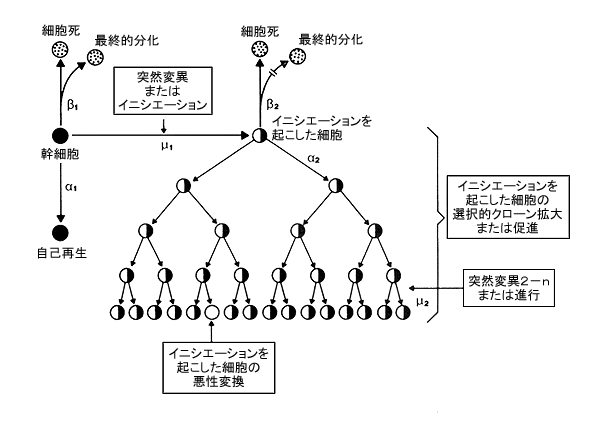
Figure 1. The initiation-promotion-progression model of carcinogenesis. Beta1, rate of terminal differentiation and death of stem cell; Beta2, rate of death, but not of terminal differentiation of the initiated cell (–||–>); Alpha1, rate of cell division of stem cells; Alpha2, rate of cell division of initiated cells; Mu1, rate of the molecular event leading to initiation (ie, possibly mutation); and Mu2, rate at which the second event occurs within an initiated cell.
If one of these promoted, initiated cells acquires additional genetic alterations (eg, other mutations, stable epigenetic changes) that allow the cell to become promoter-independent, invasive, and metastatic, then the third step of carcinogenesis–the conversion or progression stage–has occurred. This step also appears to be irreversible. Given the observations that mutagens appear to affect this stage, mutagenesis as well as stable epigenetic events could be applicable mechanisms for progression.
How do the different factors fit together?
If carcinogenesis is a multistep process, with each stage affected by different mechanisms (eg, there are many ways to cause mutations; many mechanisms lead to mitogenesis), how could a single exposure to ionizing radiation “cause” cancer? For those who harbor the idea that ionizing radiation “causes” cancer, does it not seem incredible that after an acute exposure, radiation would not only have to activate one or more oncogenes, as well as inactivate suppressor genes, it would also have to initiate, promote, or clonally expand that cell manyfold and then convert one of those initiated cells by mutating other genes to have invasive and metastatic abilities by a series of independent events in a single cell?
Would it not be more informative to ask questions such as: “Which step(s) of carcinogenesis might be affected by ionizing radiation?”; “By what mechanisms might ionizing radiation initiate, promote, or bring about progression of carcinogenesis?”; “Does ionizing radiation activate oncogenes?”; and “Does ionizing radiation deactivate tumor suppressor genes?” Moreover, does a linear-no threshold model describe the underlying mechanisms of the multistage nature of carcinogenesis, especially the promotion or mitogenic step? A recent review of chemical carcinogenic studies appears to indicate a no-effect, threshold level for the role of the mitogenic process, primarily linked to the promotion or clonal expansion of the initiated cell (S Cohen and L Ellwein, Cancer Res52:6493-505, 1991). Serious examination, as suggested in an accompanying article by VP Bond (RERF Update 4[1]:7, 1992), must be considered. By rephrasing the problem, specific testable hypotheses, using these concepts and molecular technologies, might provide new insights into how (in our case) exposure to A-bomb radiation could have contributed to the process of carcinogenesis.
Epidemiologists involved in determining the risks of radiation exposure use the term “confounding factors” for factors, such as age at exposure, time elapsed since exposure, sex, reproductive history, diet, postirradiation therapy, etc, that are known to be associated with “modifying” the radiation response. In the context of the carcinogenic process outlined above, the term “confounding factors” is very misleading. In fact, the term ought to be “contributing factors,” with radiation being only one of these factors.
Oncogenes, tumor suppressor genes, and intercellular communication
The initiation/promotion/progression hypothesis of carcinogenesis is an operational concept derived from whole-animal experiments, having no implied underlying molecular mechanism. Independently, the oncogene/tumor suppressor gene hypothesis is a concept derived empirically from molecular in vivo and in vitro studies. However, to date no viable cellular mechanism has been offered as to how the various oncogenes and tumor suppressor genes might function to convert a contact-inhibitable progenitor cell to a cancer cell, ie, one that is not contact-inhibitable and unable to terminally differentiate.
In a multicellular organism, tight regulation of a cell’s ability to proliferate and to differentiate must occur. Various intercellular communication mechanisms, such as extracellular communication via growth factors, hormones, or gap-junctional intercellular communication (GJIC) via ions and small molecules through gap-junction channels, appear to be directly associated with the regulation of cell growth and differentiation. Since the major phenotypic dysfunctions of cancer cells seem to be the lack of contact inhibition and loss of growth control and the ability to terminally differentiate, it would be reasonable to speculate that intercellular communication has been disrupted during the carcinogenic process. Indeed, many if not all cancer cells have abnormal homologous or selective communication characteristics. Many chemical tumor promoters, oncogenes, and growth factors also inhibit intercellular communication, whereas the few antitumor agents and anticarcinogens seem to up-regulate GJIC (JE Trosko et al, Pathobiology58:265-78, 1990; JE Trosko et al, Radiat Res 123:241-51, 1990). One tumor suppressor gene has been associated with a gap-junction gene (SW Lee et al, Proc Natl Acad Sci USA88:2825-9, 1991). These circumstantial, but completely independent, observations are consistent with the hypothesis that the oncogene/suppressor gene function modulates GJIC, which, in turn, modulates a cell’s ability to proliferate or differentiate.
The role of ionizing radiation in carcinogenesis
The question now arises as to how radiation might affect one or more of the mechanisms underlying the initiation, promotion, and progression phases of carcinogenesis.
Radiation: a weak initiator
The current weight of the evidence indicates that ionizing radiation is a rather weak point mutagen but a good clastogen (inducer of chromosome breaks, deletions, and rearrangements). At high enough doses, this translates into radiation being a good cytotoxicant since chromosome deletions and many types of chromosome rearrangements are lethal. In contrast, given the oncogene/tumor suppressor gene paradigm, where a balance between proto-oncogenes and tumor suppressor genes is needed for normal growth control and differentiation (see Figure 2), ionizing radiation might generally be predicted to be a rather weak “activator” of oncogenes but a strong “deactivator” of tumor suppressor genes, except possibly by causing rearrangement of normal proto-oncogenes causing them to be abnormally and stably expressed.
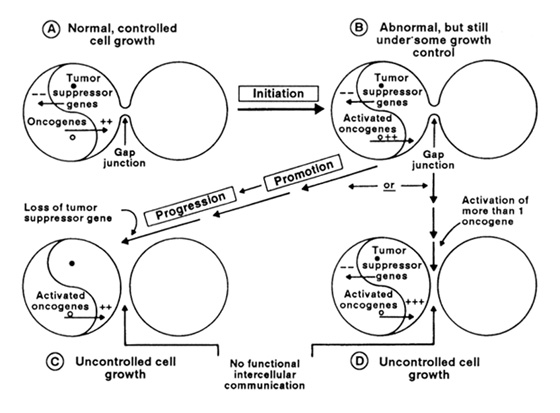
Figure 2. The yin/yang model of oncogenes and tumor suppressor genes in the control of cell growth depicts the balance between positive factors that stimulate growth and negative factors that suppress growth. In the quiescent state of a normal cell that is contact inhibited (solid tissue) or suppressed by extracellular regulators (soft tissues), the two factors balance out. During the initiation-promotion-progression process of carcinogenesis, activation of oncogenes could occur, followed by clonal expansion of these cells. The loss of tumor suppressor genes by mutation or by deletion allows the cell to enter the progression phase by stimulation of cell growth or by inability to respond to negative growth control (ie, growth inhibition). The role of gap junctional intercellular communication (for cells in solid tissues) is speculated to be down-regulated by oncogenes and up-regulated by tumor suppressor genes.
Radiation as a promoter
If ionizing radiation is to be a promoter, ie, a stimulant of the clonal expansion of initiated cells, it must be given at a high enough dose to cause significant cell killing, which, in turn, would induce compensatory hyperplasia. If one of the surviving stem cells were to have been previously initiated by some other environmental mutagen or initiated by the radiation itself, then regenerative or compensatory hyperplasia could be seen as “promotion.” If the dose were too high, the ionizing radiation would start to kill some of the preexisting or newly induced initiated cells, thereby decreasing the cancer incidence and increasing the likelihood of early death of the organism.
Radiation as a progressor
If the exposed individual has preexisting initiated and promoted clones of cells (as we all must: the older we get, the more of these we should have), then ionizing radiation, as an effective gene and chromosome deletion mutagen, might be expected to be a relatively good tumor suppressor gene “deactivator.”
Assuming that stem cells are the target cells for carcinogenesis, then the risk that the initiation step of carcinogenesis occurs from a single exposure to ionizing radiation is influenced by the number of these stem cells. Some tissues will have relatively stable numbers of these cells during aging (eg, skin and the lining of the GI tract), whereas others, such as the breast, liver, and brain tissues, may have decreasing numbers of stem cells (due to many intrinsic and extrinsic factors that I won’t discuss here).
On the other hand, depending on previous exposure to other point mutagens that might initiate (activate oncogenes) and promote (age might be an important factor here), ionizing radiation might be a good “progressor.” The interaction of sunlight exposure and ionizing radiation in skin cells of older individuals, in sun-exposed and non-sun-exposed areas, might be a test of this hypothesis, since UV radiation is a good point mutagen and skin carcinogen (presumed initiator and activator of oncogenes, promoter via its ability to kill cells, and progressor by its ability to deactivate tumor suppressor genes by point mutations).
Does radiation “cause” cancer?
Clearly, radiation is a contributing factor. How it contributes in the multistage process of carcinogenesis is not yet known; however, radiation does seem to be a weak initiator, both by its ability to induce chromosome rearrangements (to activate oncogenes) and by its weak point mutagenic potential. At higher doses, radiation might act as an indirect promoter of preexisting initiated stem cells by its ability to induce regenerative hyperplasia via its cell-killing effects. Finally, because of its ability to delete genes and chromosomes, it should be effective as a deactivator of tumor suppressor genes and therefore should act as a progressor at the late stages of carcinogenesis.
All this does not imply that ionizing radiation is unimportant in bringing about cancer. However, only by knowing the mechanisms by which ionizing radiation influences this complex disease process can we hope to develop meaningful risk estimates from studies of populations and individuals exposed to radiation, especially at low doses where the greatest uncertainties exist. Clearly, epidemiological and statistical analyses of radiation-exposed populations are critical and necessary. So are basic and fundamental studies of radiation effects on molecules, cells, and animals. In addition, the future demands a greater interaction among epidemiologists and radiation molecular and cell biologists for better hypothesis design, testing, and interpretion of epidemiologic studies. At the same time, epidemiological findings should stimulate laboratory research to explain the results. The emerging field of molecular epidemiology may provide the stimulus for this union (PG Shields and CC Harris, J Am Med Assoc 266: 681-7, 1991). At RERF, we are developing plans to move in this direction.
Note added in proof: Three recent papers (D Zhu et al, Proc Natl Acad Sci USA88:1883-7, 1991; PP Mehta et al, J Membrane Biol 124:207-25, 1991; B Ehlibali et al, Proc Natl Acad Sci USA 88:10701-5, 1991) have demonstrated that noncommunicating cancer cells, when transfected with expressible gap-junction genes, had restored cell-to-cell communication and normal growth patterns.